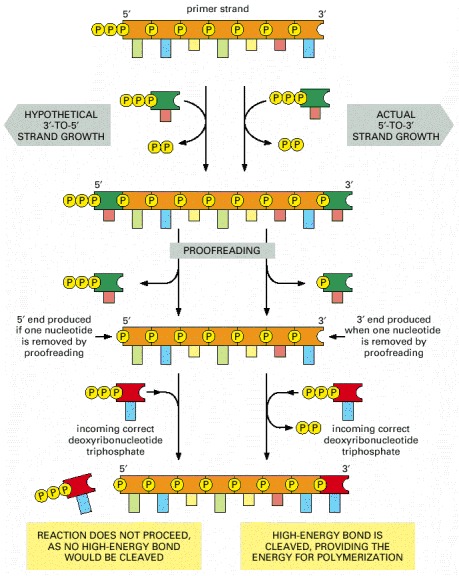
"I was going over the material and I am confused about one thing: High frequency AP's cause significant amount of glutamate release, binding to AMPA --> significant Na+ entry --> NMDAR release of Mg2+ --> --> --> LTP ... but further studies find "silent synapses" deficient in AMPA receptors do not respond to pre-synaptic activity (release of glutamate), and LTP is necessary for appearance of AMPARs. If entry of sodium via AMPARs cause LTP by causing changes in NMDAR, allowing Ca2+ entry, how can LTP occur in silent synapses? Are all synapses initially silent? or is absence of AMPA a property of only a specific subset of neurons? Does this have anything to do with backpropagating spikes?"
I answered as follows:
"Yes it does have to do with backAPs. The key point is that ltp is triggered not by local depolarisation (at the synapse itself, eg caused by locally released glu acting on ampaRs) but by the occurrence of a spike (starting at the beginning of the postsynaptic axon, the initial segment) which backpropagates into the dendrites and thus reaches all the synapses.
This answers your dilemma: at a silent synapse (most synapses start silent) the release of glu does not cause significant local depolarization, but nevertheless the neuron might fire a spike (as a result of near-simultaneous glu release at other, non-silent, synapses). When that spike backprops and reaches the silent synapse in question, it will trigger unblocking of the NMDARs at that synapse, and Ca entry, which can trigger strengthening (by adding AMPARs).
The whole point here that the decision to strengthen a particular synapse (or not) should depend not just on what is happening at that synapse (i.e. the arrival of a presynaptic spike) but the collective decision of the whole postsynaptic neuron (does it fire a spike too?).
Similarly at a particular nonsilent synapse: this synapse will depolarise (because it already has some ampaRs) if the input axon fires, but we don't want that synapse to locally depolarise enough to unblock the nmdars because then it would strengthen regardless of what the whole neuron is doing - we only want the synapse to strengthen if the postsynaptic neuron fires (mostly as a result of the other active synapses, but in small part because of the synapse in question). So we can adjust each synapse individually based on what the relevant pre- and postsynaptic axon are doing: if they both fire (the pre slightly before the post) then strengthen it! This mechanism implements the Hebb rule, fire together wire together.
We can capture the behavior of a neuron with 2 simple equations: dw/dt = xy and y = f(x.w). In the first equation w refers to the strength of a particular synapse (it should have a subscript i since we are referring to the ith synapse) and y is the firing rate of the neuron. The second, "dot-product", equation says y depends on how well the current input pattern (vector x) matches the current strength pattern of the whole set of synapses (the vector w). The 2 equations together mean that gradually over time the relative strength of all the synapses will come to reflect regularities (correlations) in the entire set of input pattens the neuron sees over its lifetime (which is essentially the lifetime of the animal). Since detecting regularities is what we mean by "understanding", this implies the brain (just a collection of neurons!) can "understand" - i.e. have a mind (a not uninteresting conclusion).
Of course the devil is in the details. The whole thing would be undermined if (a) the local depolarisation due to ampaRs were big enough to unblock the nmdaRs or (b) the Ca signal at one synapse could influence, even to a tiny degree, what happens at other synapses. Both these point are controversial.
In the case where all the synapses are initially silent (eg in the early fetus), it would appear that there's no way to get the ball rolling: no ampars means no firing! However, it turns out that very early on GABA acts as an excitatory transmitter (chloride pumps have not yet matured and Ecl is positive to threshold)! Probably initially random firing leads to some random unsilencing, and only later do the synapses get further adjusted in an experience-dependent way.
If you can answer the question and my answer, the course will have been a success, no matter what your final grade.